Molecules containing simple alkene functionality are attractive substrates in synthesis. Carbon-carbon double bonds are relatively stable groups, lacking polarisation, they can be carried through synthetic processes involving acid, base, mildly oxidative and reductive conditions. But, the availability of the π-electrons for reaction embodies remarkable potential for constructing substituted contiguous stereocentres and the possibility of introducing complex functionality to an all-carbon system.
In recent years, functionalisation of unactivated alkenes has blossomed with developments in robust metal-catalysed processes and new oxidating agents. In the last few weeks several reports of new alkene functionalisations have been published that cover many aspects of this broad area, and show the diverse utility of alkenes in synthesis.
Hayashi and co-workers have published an elegant cyclising difunctionalisation of dienes.[1] The process is an iridium-catalysed C-H activation of cyclic aryl N-sulfonyl ketimines and gives spirocyclic aminoindane scaffolds with high diastereoselectivity.

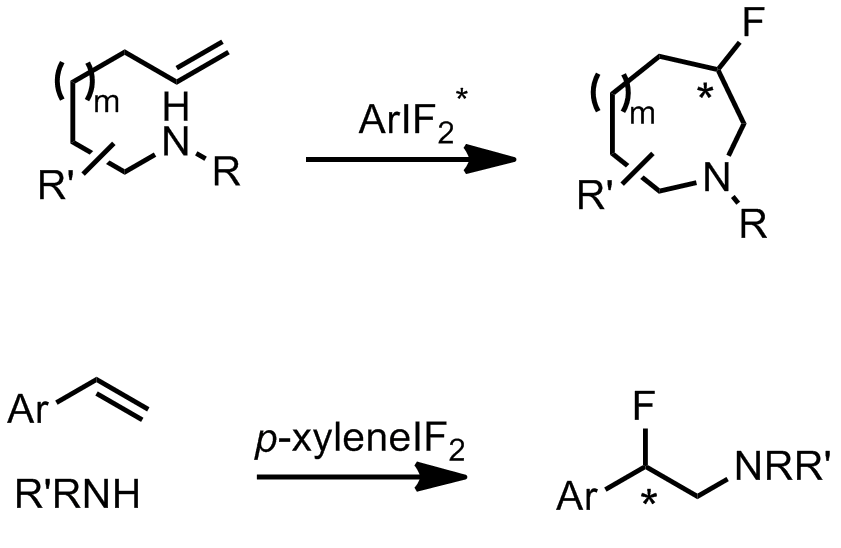
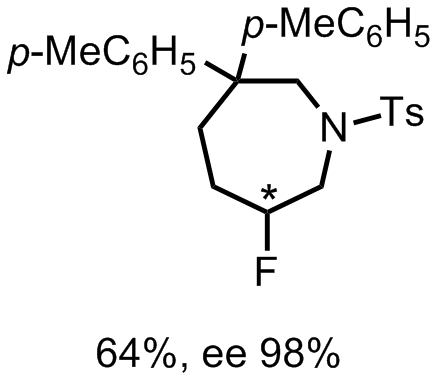
We’ve already discussed an asymmetric cyclising aminofluorination reaction carried out by a chiral hypervalent iodine fluoride oxidant.[2] The reaction is endo-selective and prepares fluoropiperidine-based systems. Extension of the reaction to a racemic intermolecular process with styrene substrates was also demonstrated.

In a similar example of intermolecular alkene difunctionalisation, Zhang reports aminocyanation and diamination of (predominantly) styrene substrates.[3] The principle reagent for functionalisation is NFSI (N-fluorobenzenesulfonimide), which aminates the terminal end of the double bond. Subsequent reaction with TMSCN gives the aminonitrile product. Alternatively, an alkylnitrile may be used to give the Ritter product of the second addition.

The authors suggest a copper(I)-catalysed radical mechanism, generating a carbon-centered radical intermediate after amination by •N(SO2Ph)2. This reaction gives some idea of the kind of complex functionality that can be introduced to all-carbon alkene substrates by oxidative processes.
Hydrofunctionalisation of alkenes doesn’t provide the same high degree of complexity as oxidative difunctionalisation processes, but reactions in this category do offer a great deal of diversity in synthetic options while still providing regio- and stereocontrol of sp3 carbon centres. A report from Qing and co-workers describes a general and high yielding hydrotrifluoromethylation of unactivated alkenes.[4] Trifluoromethyl groups are useful moieties in pharmaceutical development of lead compounds. The group apply the method to a wide scope of unactivated alkene substrates showing tolerance to many other functionalities.

Hartwig’s group in Berkeley have developed an asymmetric hydroheteroarylation of bicycloalkenes.[5] The reaction is catalysed by an iridium DTBM-Segphos complex and tolerates various heteroaromatic coupling partners.

Alkenes offer chemists widely available, stable and highly tolerant substrates for synthesis. Careful application of modern reactions allows the uncovering of diverse and complex functionality from a carbon-carbon double bond, and new developments provide ever more effective methods for the preparation of desirable adducts.
References:
1. DOI: 10.1021/ja311968d
2. DOI: 10.1002/anie.201208471
3. DOI: 10.1002/anie.201209142
4. DOI: 10.1002/anie.201208971
5. DOI: 10.1021/ja312360c