Wangqing Kong, Pascal Feige, Teresa de Haro, and Cristina Nevado
Angew. Chem. Int. Ed.
DOI: 10.1002/anie.201208471
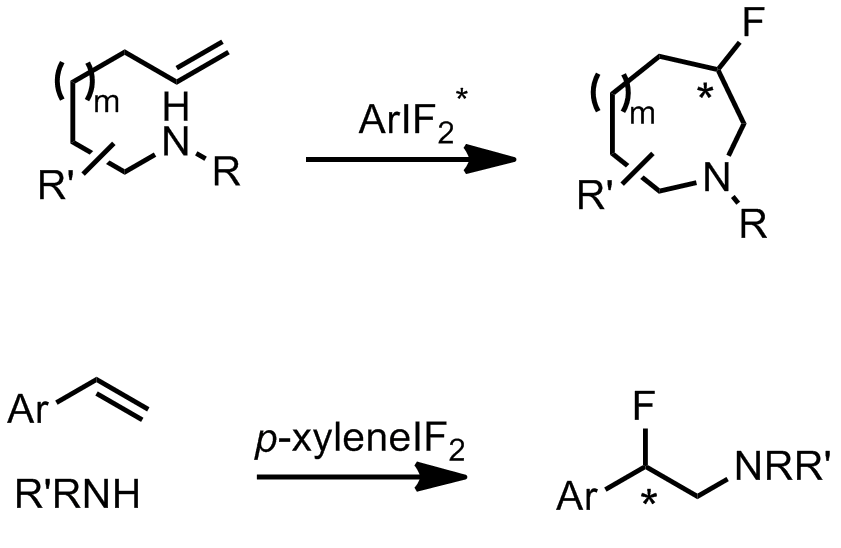
Multisubstituded, saturated 5-, 6- and 7- membered heterocycles are part of many bio-active molecules. Fluorinated derivatives of such structures have also been shown to display better pharmacological properties such as solubility and metabolic stability. Hence their enantioselective preparation in a facile and efficient manner is of great interest for synthetic chemists.

Classical approaches involve cycloadditions such as inverse-electron demand Diels-Alder or [2,3] azomethine ylide reactions1, whereas more modern approaches focus on transition metal catalysed aminoalkylations2. Cristina Nevado’s group in Zurich, have recently reported a metal-free regio- and enantioselective aminofluorination for the preparation of 6- and 7-membered fluorinated heterocyclic compounds.

The reaction is highly regioselective yielding the 6-endo-cyclisation product only. Using their newly discovered conditions they investigated the scope of the intramolecular aminofluorination.

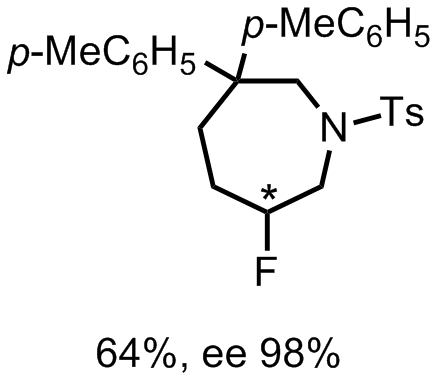
From the data presented in the supporting information, aromatic substituents seem to have a positive influence on enantioselective induction. The preparation of 7-membered rings was also performed. However it required the addition of a catalytic amount of [(Ph3P)AuNTf2].
The authors also investigated the scope of intermolecular aminofluorinations in a non-asymmetric manner, using p-xyleneIF2 as the fluorine source.

This method provides a facile route to fluorinated surrogates of saturated heterocyclic compounds. It would be interesting to see it applied for the preparation of morpholine or piperazine containing compounds in the future.
References:
1. a) Geraldine Masson et al. Org. Bio. Chem. 2012 ; b) Marco Potowski et al. Angew. Chem. Int. Ed. 2012.
2. a) Josephine S. Nakhla et al. Org. Let. 2007 ; b) Matthew L. Leathen et al. J. Org. Chem. 2009.




























