Robert W. Huigens III, Karen C. Morrison, Robert W. Hicklin, Timothy A. Flood Jr, Michelle F. Richter and Paul J. Hergenrother
Nature Chem.
DOI: 10.1038/nchem.1549
To synthetic chemists, natural products are generally viewed as end-points in synthetic projects (or occasionally as tools, such as ligands or catalysts). Rarely, are complex natural products considered as launching-off points in the synthesis of other interesting molecules. A recent report takes this rather unusual approach by applying the principles of diversity oriented synthesis (DOS) in preparing a series of molecules with drug-like molecular properties.
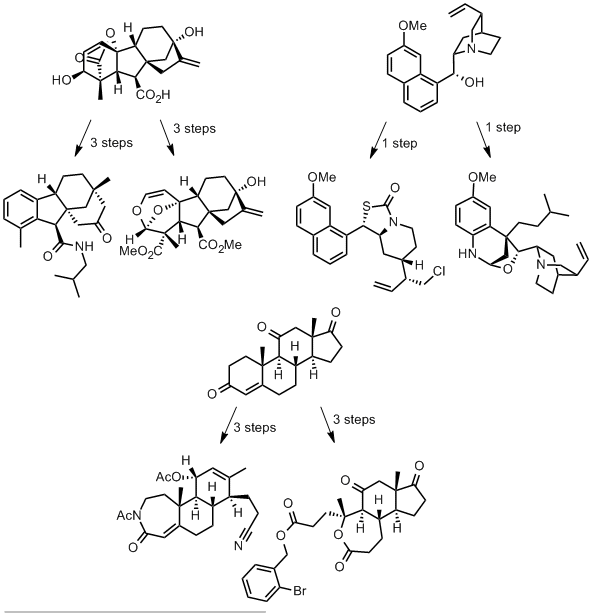
The authors take three widely available natural products and carry out numerous derivatisations and transformations to give 49 dissimilar molecular scaffolds, a process they dub ‘compexity-to-diversity’ (CtD). A few examples are shown below.
These molecules are presented as exemplifying a new approach to preparing drug-like molecules. They are analysed in characteristics typical of drug molecules and compared with a common screening library of 150,000 molecules.
In terms of diversity, these molecules display considerable structural dissimilarity (established by calculating Tanimoto coefficients for each pair of molecules), even within derivatives of the same natural product.
The CtD molecules display superior lipophilicity (average ClogP = 2.90) compared with the screening collection (average ClogP = 3.99), with over 60% of CtD molecules in the optimal logP range of 0 to 4. Similarly, the CtD library also displays a far higher fraction of sp3 character than the screening compounds, a property indicating a high degree of 3D structure and correlated with enhanced aqueous solubility.
The authors argue that molecular complexity is advantageous in drug-like molecules as more complex molecules might bind their targets more specifically. They point to the number of stereogenic centres the molecule contains as a surrogate for molecular complexity and show that their CtD molecules contain far more stereocentres than the compounds in the screening collection.
Despite demonstrating clear drug-like properties (particularly ClogP), the molecules have not been analysed in another significant property indicative of the likelihood of observing drug-like behaviour: molecular weight. While molecular complexity offers the possibility of tighter target binding, it also decreases the chances of observing binding in any given target site. This is correlated with molecular weight, where it is estimated that each heavy atom added to the molecule increases the number of potential structures by a factor of 10.1 The chances of finding hit molecules is increased when screening molecules with lower molecular weight (due to better sampling of chemical space), albeit with potentially weaker binding (cf. fragment-based design).
The six CtD molecules displayed above lie in the molecular weight range of 353 to 505, outside of the optimal range of 200 to 350 for lead compounds, though within the boundaries of the Lipinski rule of 5 for drug-like molecules.
This approach is, of course, not limited to the derivatisation of the natural products or classes presented within this paper, but that similar structural modifications may be carried out on any readily available complex molecules to prepare diverse structures with drug-like properties.
1. A. Nadin, C. Hattotuwagama, I. Churcher Angew. Chem. Int. Ed. 2012, 51, 1114 and references 27-29 therein.